Image


Postdoc: Dana-Farber Cancer Institute, Harvard Medical School
Ph.D.: Washington University in St. Louis
B.S.: Seoul National University, South Korea
DNA damage repair and cancer therapeutics
| Our laboratory aims to elucidate the fundamental mechanisms of DNA repair with the goal of developing anti-cancer targets and biomarkers for therapeutic benefit. Genome instability, defined by a high frequency of DNA mutations primarily caused by defective DNA repair, is one of the most pervasive characteristics of cancer, contributing to the pathogenesis of both hereditary and sporadic cancers. The DNA repair system functions as a critical tumor suppressor network that preserves genomic integrity, thereby preventing the onset and progression of tumors. Therapeutic interventions that exploit dysregulated DNA repair in cancer can induce synthetic lethality (exemplified by the FDA approval of PARP inhibitors to treat advanced ovarian cancer with defective BRCA1/2 genes) or augment the efficacy of cytotoxic chemotherapy. Thus, understanding the regulatory networks of DNA repair will not only provide insights into cancer etiology driven by genome instability, but also facilitate the development of novel cancer therapeutics. Using cellular, biochemical, and genetic approaches, we are investigating mechanisms of genome maintenance at DNA replication forks and pathways of genotoxicity triggered by anti-cancer therapies. | 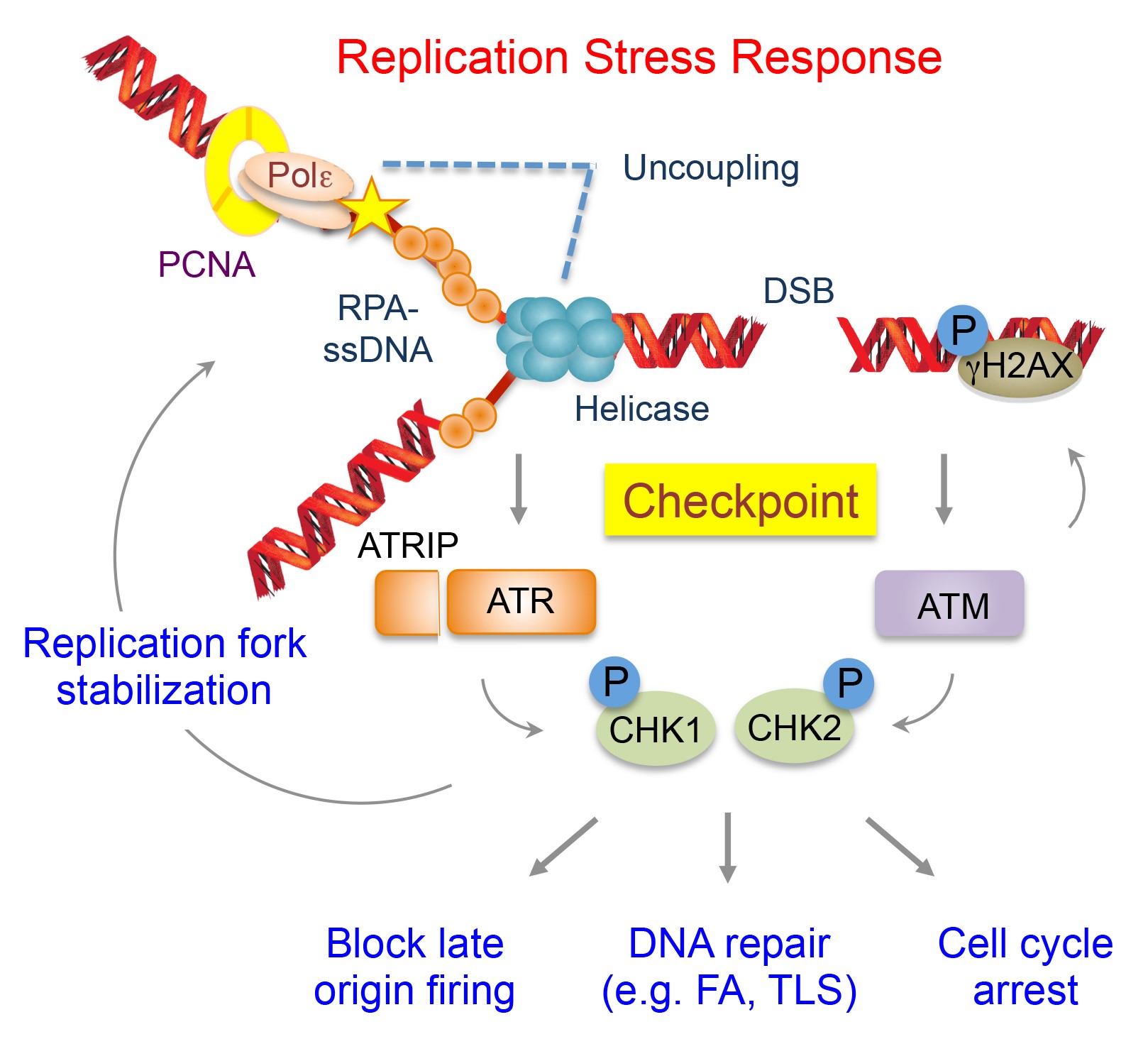 |
1. Mechanisms of the DNA replications stress response
Achieving accurate genome duplication is a challenging task for the DNA replication machinery. Failure to overcome various replication-blocking obstacles causes perturbations of DNA synthesis, collectively referred to as DNA replication stress. Increased formation of aberrant replication fork structures due to the loss of the replication stress response pathways leads to deleterious mutational events that are associated with genome instability and tumorigenesis. As such, elevated DNA replication stress is prevalent in cancer. However, this also provides a therapeutic opportunity where we can exacerbate the replication stress of cancer cells to achieve specific killing of cancer cells. Our research focus on defining the regulatory axis of the DNA replication stress signaling orchestrated by the DNA damage response kinases and the DNA replication machinery, including TIMELESS (TIM)-TIPIN in the fork protection complex (FPC) and other emerging players at DNA replication forks. Fundamental knowledge on the DNA replication and stress responses will provide opportunities to exploit the DNA replication vulnerabilities of cancer cells for therapy.
2. Exploiting DNA replication vulnerability for cancer therapy
Exacerbating replication-associated DNA lesions is the outcome of many traditional cytotoxic chemotherapies and DNA replication checkpoint inhibitors, including topoisomerase inhibitors, ATR inhibitors, and CHK1 inhibitors, as well as PARP inhibitors specific for BRCA1/2 mutations. However, they often induce a reversible form of growth arrest known as therapy-induced senescence (TIS). TIS promotes drug resistance and stemness, while the senescence-associated secretory phenotype (SASP) creates a proinflammatory niche for aggressive tumor progression. Because TIS is reversible, cells can potentially be reprogrammed toward cell death. We previously showed that TIM degradation synergizes with ATR inhibition to trigger DNA replication catastrophe, shifting cell fate from TIS to death. We are interested in elucidating the molecular "decision" between survival and death; Knowledge from this study will help develop rational drug combinations or new strategies to prevent tumor dormancy and render cells more responsive to conventional or targeted therapies.
3. Pathways of oncogene-induced senescence and transformation
In early lesions, oncogenic stimuli like RAS trigger oncogene-induced senescence (OIS) as a barrier to malignancy. Establishing OIS depends on a sustained DNA damage response arising from replication stress. Malignant progression requires escaping senescence and inactivating the DNA damage response, accelerating carcinogenesis via genome instability. However, how oncogenic signaling impairs replication fork integrity and disrupts protection mechanisms, such as fork reversal, remains unclear. We are investigating this link between oncogenic signaling and fork protection using HRAS and KRAS prostate cancer organoid/mouse models.
Hyungjin Kim, Ph.D.
Professor
Department of Pharmacological Sciences
Renaissance School of Medicine and the Cancer Center
Basic Sciences Tower 8-125
Stony Brook University
Office: 631-444-3134